
Biochemistry is one of the Crucial subject for NEET PG . It accounts for around 10 – 15 % of the Total marks . Compared to other first year MBBS subjects , Biochemistry is considered as more Scoring. Fundamental Concepts remains Constant, making it less prone to surprises. It is also Considered as Foundation for Medicine where the understanding of Biochemical processes forms the bedrock for comprehending various physiological functions and diseases.
1: A patient with deafness and mental retardation is found to have an excess accumulation of glycosaminoglycans in his body. After a detailed evaluation, he is diagnosed with Hunter disease. What is the site of synthesis of these accumulated substances?
a: Mitochondria and Golgi apparatus
b: Endoplasmic reticulum and Golgi apparatus
c: Lysosomes and endoplasmic reticulum
d: Endoplasmic reticulum and mitochondria
Solution: Correct answer is ‘b. i.e., Endoplasmic reticulum and Golgi apparatus
The sites of synthesis of glycosaminoglycans (GAG) are the endoplasmic reticulum and Golgi apparatus.
Synthesis of GAGs:
1: Synthesis of the core proteins: Endoplasmic reticulum
2: Biosynthesis of glycosaminoglycan chains and their subsequent modifications: Golgi apparatus
Glycosaminoglycans or mucopolysaccharides are complex carbohydrates made up of amino sugars and uronic acids. They may be attached to a protein molecule to form a proteoglycan. Proteoglycans are components of the ground substance of connective tissue. Proteoglycans have a bottle brush appearance.
The biochemically important GAGs are as follows:
1: Hyaluronic acid
2: Chondroitin sulfate
3: Keratan sulfate
4: Heparin
5: Heparan sulfate
6: Dermatan sulfate
Hunter disease, also known as mucopolysaccharidosis type II, is a rare genetic disorder leading to the accumulation of glycosaminoglycans in the body. GAGs are degraded by lysosomal enzymes, which are deficient in this condition.
Note: Hyaluronic acid is the only GAG that is not synthesized by the Golgi apparatus. It is formed at the plasma membrane.
2: A 5 year old mentally retarded girl presents with a protuberant abdomen, short stature, coarse facial features, and cloudy corneas. Skeletal malformations include dysostosis multiplex and bullet-shaped middle phalanx. Which of the following enzyme deficiencies is most likely in this patient?
a: Iduronate sulfatase
b: β- Galactosidase
c: α-L- Iduronidase
d: β-Glucuronidase
Solution: Correct answer is ‘c’ i.e., α-L- Iduronidase
The clinical features point to the diagnosis of Hurler’s disease (MPS 1). Deficiency of the enzyme α-L-iduronidase that degrades glycosaminoglycans results in Hurler’s disease.
Hurler’s disease was also known previously as gargoylism (coarse facial features)
Clinical features of the disease include:
1:Coarse facial features
2:Corneal clouding
3: Large tongue
4:Inguinal hernia
5: Hepatosplenomegaly
6: Joint stiffness
7: Short stature
8: Copious nasal discharge
9: Skeletal dysplasia
10: Hydrocephalus
11: Valvular heart disease
12: Reilly body inclusions are present in leucocytes
The gene encoding α-L-iduronidase is present on chromosome 4. Heparan sulfate and dermatan sulfate are accumulated.
Enzyme replacement therapy is available in the form of aldurazyme.
Mucopolysaccharidoses are a group of metabolic disorders caused by the absence or malfunctioning of lysosomal enzymes needed to break down molecules called glycosaminoglycans.
- Most common MPS: Sanfilippo > Hunter’s and Hurler’s
- MPS with no mental retardation: Scheie’s disease, Morquio syndrome, and Maroteaux – Lamy syndrome.
- MPS with no corneal clouding: Hunter’s disease and Sanfilippo syndrome.
- MPS with XLR inheritance: Hunter’s disease
- MPS with no visceromegaly: Morquio’s disease
- Skeletal deformity associated with all MPS: Dysostosis multiplex
- MPS with no leucocyte inclusions: Morquio’s disease.
3: An adolescent male patient presented with pain in the calf muscles on exercise. On biopsy, an excessive amount of glycogen was found to be present in the muscle. What is the most likely enzyme deficiency?
a: Muscle-debranching enzyme
b: Phosphofructokinase 1
c: Glucose 6 phosphatase
d: Muscle glycogen phosphorylase
Solution: Correct answer is ‘d’ i.e., Muscle glycogen phosphorylase
The given clinical features are seen in McArdle’s disease. This patient has muscle glycogen phosphorylase deficiency.
The lack of muscle glycogen phosphorylase results in excess glycogen build-up in muscles.
Clinical features include:
1: Poor exercise tolerance
2: Abnormally high muscle glycogen (2.5-4%)
3: Very low blood lactate after exercise
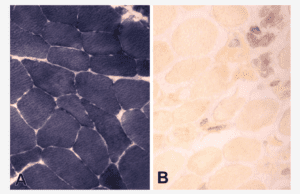
The images given below show myophosphorylase enzyme histochemistry showing (A) normal muscle and (B) McArdles’ disease.
4: Which of the following enzymes catalyze the rate – limiting step of glycogenolysis?
a: Glycogen synthase
b: Glycogen phosphorylase
c: Glucose 6 Phosphatase
d: Phosphoglucomutase
Solution: Correct answer is ‘b’ i.e., Glycogen phosphorylase
The rate – limiting step in glycogenolysis is catalyzed by the enzyme, glycogen phosphorylase.
The process of phosphorolytic cleavage of the α1 à 4 linkages of glycogen to yield single units of glycose 1-phosphate, is catalyzed by the enzyme glycogen phosphorylase.
Glycogen phosphorylase requires pyridoxal phosphate as the coenzyme. The phosphate group is catalytically active. There are different isoenzymes of glycogen phosphorylase in the liver, muscle, and brain.
5: A neonate presents with feeding difficulty and failure to thrive. Her urine has a peculiar burnt sugar odor due to an excess of branched – chain amino acids. Which of the following is unlikely to be elevated?
a: Valine
b: Isoleucine
c: Leucine
d: Lysine
Solution: Correct answer is ‘d’ i.e., Lysine
The given clinical scenario is suggestive of maple syrup urine disease (MSUD). Lysine is not a branched-chain amino acid and hence is unlikely to be elevated.
Branched – chain amino acids are:
1: Valine (V)
2: Isoleucine (I)
3: Leucine (L)
MSUD is due to the deficiency of branched – chain alpha – keto acid dehydrogenase complex (BCKDC), leading to the formation of branched – chain amino acids and their toxic by-products (ketoacids) in the blood and urine. There is a peculiar odor of burnt sugar found in urine, sweat, and cerumen
6: A forensic team used performed a ninhydrin test on some samples obtained from a crime scene. This test can detect which of the following?
a: Bile salts
b: Amino acids
c: Nucleic acids
d: Lipids
Solution: Correct answer is ‘d’ i.e., Lipids
Ninhydrin test is used to detect amino acids.
A purple – colored complex (Ruhemann’s purple) indicates a positive reaction. Proline and hydroxyproline form a yellow adduct with ninhydrin. Glutamine and asparagine produce a brown color.
This test is used effectively used at crime scenes to develop and identify latent fingerprints.
| Reactions | Specific Group or Amino Acid |
| Biuret Reaction | Two peptide linkages |
| Ninhydrin Reaction | Alpha – amino acids |
| Xanthoproteic Reaction | Benzene ring of aromatic amino acids |
| Millons Reaction | Phenolic group (Tyrosine) |
| Hopkins-Cole Reaction | Indole ring (Tryptophan) |
| Sakaguchi Reaction | Guanidine group (Arginine) |
| Nitroprusside Reaction | Sulfhydryl groups (Cysteine) |
| Sulfur Test | Sulfhydryl groups (Cysteine) |
| Pauly’s Test | Imidazole ring (Histidine) |
| Folin-Ciocalteu’s Test | Phenolic group (Tyrosine) |
7: A 43 year old man presented with intermittent upper abdominal pain for 4 months, episodic hot flushes involving the face and upper chest for 2 months, and watery stools 5 – 6 episodes per day for 1 month. Urinalysis was positive for 5 – OH indole acetic acid. Which of the following is responsible for these symptoms in him?
a: 5 hydroxytryptamine
b: 5 hydroxytryptophan
c: 5 Carboxytryptamine
d: 5 Carboxytryptophan
Solution: Correct answer is ‘a’ i.e., 5 hydroxytryptamine
The given clinical scenario is suggestive of carcinoid syndrome. Excessive levels of serotonin, also known as 5 hydroxy-tryptamine is responsible for it.
Serotonin is a monoamine neurotransmitter. It is produced by hydroxylation and subsequent decarboxylation of tryptophan. It is a potent vasoconstrictor and stimulator of smooth muscle contraction.
In carcinoid syndrome, excessive levels of serotonin is produced which causes flushing, diarrhea, wheezing, damage to the tricuspid valve, etc. The levels of 5-OH indole acetic acid (serotonin metabolite) are elevated in the urine.
8: A 7 year old child is brought to the OPD with a history of impaired vision and severe photophobia. He has achieved all the developmental milestones at the appropriate age. On examination, he has fair hair, light skin color, and pale blue eyes. Which enzyme is most likely deficient in this case?
a: Tyrosinase
b: Tyrosine hydroxylase
c: Tyrosine transaminase
d: Phenylalanine hydroxylase
Solution: Correct answer is ‘a’ i.e., Tyrosinase
The clinical scenario is suggestive of albinism which occurs due to a deficiency of copper-containing enzyme tyrosinase causing a defect in melanin production.
Oculocutaneous albinism (OCA) is a group of inherited disorders characterized by a reduction or complete lack of melanin pigment in the skin, hair, and eyes. They have an increased risk for skin cancer.
Its symptoms are albinism with vision problems, which include:
- Strabismus
- Photophobia
- Nystagmus
- Impaired vision or blindness
- Astigmatism
Neuropsychiatric manifestations are not seen.
| Disorder | Enzyme defect |
| Phenylketonuria | Phenylalanine hydroxylase |
| Tyrosinosis (Tyrosinemia type I) | Maleyl Acetate isomerase or Fumarylacetoacetate hydrolase |
| Maple syrup urine disease | Branched – chain alpha – keto acid dehydrogenase (BCKAD) |
| Alkaptonuria | Homogentisate oxidase |
| Albinism | Tyrosinase |
| Homocystinuria type I | Cystathionine synthase |
| Homocystinuria type II | Defect in methylcobalamin formation |
| Cystathioninuria | Cystathionase |
| Tyrosinemia type II or Richner – Hanhart syndrome | Tyrosine transaminase or Tyrosine aminotransferase |
| Primary hyperoxaluria | Alanine glyoxalate aminotransferase |
| Hypervalinemia | Valine transaminase |
| Isovaleric acidemia | Isovaleryl CoA dehydrogenase |
| Histidinemia | Histidase |
| Hyperprolinemia type I | Proline oxidase |
9: Which of the following is responsible for the negative charge in fibrinopeptide A?
a: Glutamate and Valine
b: Histidine and Lysine
c: Aspartate and glutamate
d: Serine and Threonine
Solution: Correct answer is ‘c’ i.e., Aspartate and glutamate
Aspartate and glutamate are responsible for the negative charge in fibrinopeptide A and B of fibrinogen.
The negative charges contribute to the solubility of fibrinogen in plasma and importantly also serve to prevent aggregation by causing electrostatic repulsion between fibrinogen molecules.
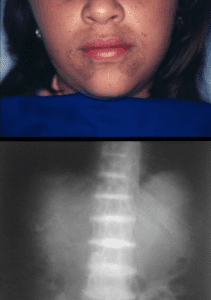
10 : The patient presents with pigmentation of the face as shown in the image below and gradually increasing backache and stiffness. The X-ray image of the spine is shown. What is the most probable diagnosis?
a: Phenylketonuria
b: Alkaptonuria
c: Maple syrup urine disease
d: Tyrosinemia
Solution: Correct answer is ‘b’ i.e., Alkaptonuria
The clinical image shows the accumulation of alkapton bodies in connective tissues on the face which is associated with alkaptonuria.
X-ray shows calcification of intervertebral discs called as ochronotic spondyloarthropathy also seen in alkaptonuria.
Alkaptonuria due to the deficiency of homogentisic acid oxidase results in the accumulation of homogentisic acid.
The condition has three characteristic symptoms:
1: Black ochronotic pigmentation (Alkapton bodies) of cartilage and collagenous tissue
2: Homogentisic aciduria (patient’s urine contains elevated levels of homogentisic acid, which is oxidized to a dark pigment on standing)
3: Large joint arthritis
| Disorder | Enzyme defect |
| Phenylketonuria | Phenylalanine hydroxylase |
| Tyrosinosis (Tyrosinemia type I) | Maleyl Acetate isomerase or Fumarylacetoacetate hydrolase |
| Maple syrup urine disease | Branched – chain alpha – keto acid dehydrogenase (BCKAD) |
| Alkaptonuria | Homogentisate oxidase |
| Albinism | Tyrosinase |
| Homocystinuria type I | Cystathionine synthase |
| Homocystinuria type II | Defect in methylcobalamin formation |
| Cystathioninuria | Cystathionase |
| Tyrosinemia type II or Richner – Hanhart syndrome | Tyrosine transaminase or Tyrosine aminotransferase |
| Primary hyperoxaluria | Alanine glyoxalate aminotransferase |
| Hypervalinemia | Valine transaminase |
| Isovaleric acidemia | Isovaleryl CoA dehydrogenase |
| Histidinemia | Histidase |
| Hyperprolinemia type I | Proline oxidase |
11: A 15 year old boy with progressive myopia and night blindness was found to have gyrate atrophy of the retina and choroid. Which of the following will be elevated in plasma?
a: Cystine
b: Ornithine
c: Lysine
d: Alanine
Solution: Correct answer is ‘b’ i.e., Ornithine
Plasma levels of ornithine are elevated in gyrate atrophy of the retina and choroid.
The initial reactions in arginine catabolism are the conversion of arginine to ornithine followed by the transamination of ornithine. Any mutations in ornithine aminotransferase lead to elevated plasma and urinary concentrations of ornithine.
This is associated with a condition known as gyrate atrophy of the choroid and retina. It presents with myopia and night blindness in adolescence that progresses to legal blindness by 50 years. The condition is treated with dietary restriction of arginine.
12: A 2 month old infant presented with abdominal distention and increased irritability for 10 days. Physical examination showed jaundice, hepatomegaly and a boiled cabbage odour. Investigations were notable for elevated transaminases and succinylacetone. What is the treatment of choice?
a: Dietary restriction of phenylalanine
b: Dietary restriction of tyrosine
c: Nitisinone (NTBC)
d: All of the above
Solution: Correct answer is option ‘d’ i.e., All of the above
The given clinical scenario is suggestive of type 1 tyrosinemia (tyrosinosis). It is treated by a combination of nitisinone and dietary restriction of phenylalanine and tyrosine.
Type 1 tyrosinemia is the most common form of tyrosinemia caused by defective fumarylacetoacetate hydrolase enzyme. They usually present between 2-6 months of age with acute liver failure. They may also present with peripheral neuropathy resembling acute porphyria. Renal involvement is manifested as a Fanconi-like syndrome with hyperphosphaturia, hypophosphatemia and vitamin D resistant rickets.
An odour resembling boiled cabbage resulting from increased methionine metabolites can be noted. Elevated levels of succinylacetone in blood or urine is diagnostic of type 1 tyrosinemia.
Note: A boiled cabbage odour can also occur in hypermethioninemia.
13: Which of the following enzymes is involved in the process of oxidative deamination?
a: Glutamate synthetase
b: Glutaminase
c: Glutamate dehydrogenase
d: Glutamate aminotransferase
Solution: Correct answer is ‘c’ i.e., Glutamate dehydrogenase
Glutamate dehydrogenase is involved in the process of oxidative deamination. In contrast to transamination (transfer amino groups), oxidative deamination by glutamate dehydrogenase results in the liberation of the amino group as free ammonia.
Glutamate dehydrogenase can use NADH or NADPH during oxidative deamination.
14: A 10 year old boy rapidly develops hypoglycemia after moderate activity. Blood examination reveals raised levels of ketone bodies, lactic acid, and triglycerides. On examination, the liver and kidneys are found to be enlarged. Histopathology of the liver shows deposits of glycogen in an excess amount. What is the diagnosis?
a: von Gierke’s disease
b: Pompe’s disease
c: McArdle’s disease
d: Cori’s disease
Solution: Correct answer is ‘a’ i.e., von Gierke’s disease
The given features are consistent with von Gierke’s disease. It is the most common glycogen storage disease.
Enzyme deficient in this condition is glucose – 6 – phosphatase (common to both glycogenolysis and gluconeogenesis). Inheritance is autosomal recessive.
Glycogen accumulation occurs in the liver and renal tubular cells. Clinical features include:
1: Severe hypoglycemia
2: Ketosis
3: Lactic acidosis
4: Hypertriglyceridemia
5: Hyperuricemia
A definitive diagnosis is made using a liver biopsy. Fructose and galactose restriction in the diet is advised.
Glycogen Storage Disorders

15: A 8 year old boy presented with dwarfism and skeletal abnormalities. Upon examination, the abdomen was distended with no hepatosplenomegaly. The child was assessed and was found to be of normal intelligence. Which of the following mucopolysaccharidoses could be a likely cause?
a: Hurler’s disease
b: Hunter’s disease
c: Sanfilippo disease
d: Morquio’s disease
Solution: Correct answer is ‘d’ i.e., Morquio’s disease
The key features in the above scenario are the absence of hepatosplenomegaly and the preservation of intelligence. Both of these features make the diagnosis of Morquio’s disease most likely. All the other conditions above are associated with hepatosplenomegaly.
Morquio disease (MPS-IV) is caused by a deficiency of N-acetylgalactosamine – 6-sulfatase (MPS-IVA) or of β-galactosidase (MPS IVB). Both lead to defective degradation of keratin sulfate.
Early symptoms:
1: Genu valgus
2: Kyphosis
3: Growth retardation with short trunk and neck
4: Waddling gait
5: Preservation of intelligence
6: Instability of the odontoid process and ligamentous laxity is regularly present and can result in life-threatening atlantoaxial instability and dislocation.
It is of two types:
| Type | Enzyme involved | Chromosome |
| Morquio syndrome A | Galactosamine 6-sulfatase | Chromosome 16 |
| Morquio syndrome B | β-Galactosidase | Chromosome 4 |
Note:
- MPS with no mental retardation: Scheie’s disease, Morquio syndrome, and Maroteaux – Lamy syndrome.
- MPS with no visceromegaly: Morquio’s disease
16: You are collecting a blood sample from a patient for blood glucose estimation. You put the sample in a vacutainer containing sodium fluoride. Which of the following enzyme is inhibited by this compound?
a: Glyceraldehyde 3 PO4 dehydrogenase
b: Enolase
c: Pyruvate kinase
d: 1, 3 bisphosphoglycerate kinase
Solution: Correct answer is ‘b’ i.e., Enolase
Sodium fluoride and potassium oxalate are added to the blood sample to estimate glucose. Fluoride is an inhibitor of enolase and hence halts the process of glycolysis. By doing so, it prevents blood glucose levels from being falsely estimated as low.
Inhibitors of glycolytic enzymes:
1: Fluoride inhibits enolase
2: Iodoacetate inhibits glyceraldehyde 3 PO4 dehydrogenase
3: Arsenate inhibits phosphoglycerate kinase
Inhibitors of glycolysis
| Agent | Enzyme |
| Iodoacetate | Glyceraldehyde 3 – phosphate dehydrogenase |
| Arsenate | Phosphoglycerate kinase |
| Fluoride | Enolase |
Inhibitors of TCA cycle
| Agent | Enzyme |
| Fluoroacetate | Aconitase |
| Arsenite | α – ketoglutarate dehydrogenase |
| Malonate | Succinate dehydrogenase |
Inhibitors of electron transport
| Complex | Inhibitors |
| Complex I | Piercidin, Amobarbital, Rotenone |
| Complex II | TTFA, Carboxin, Malonate |
| Complex III | BAL, Antimycin A |
| Complex IV | Hydrogen sulphide, Carbon monoxide, Cyanide |
Inhibitors of oxidative phosphorylation
| Agent | Mechanism |
| Atractyloside | Inhibits ADP and ATP Transporter |
| Oligomycin | Blocks flow of protons through Fo complex |
17 : What is the process of reconversion of lactate formed in the muscles and RBC’s back into glucose inside the liver called?
a: Cahill cycle
b: Cori cycle
c: Kreb’s cycle
d: Dicken – Horecker cycle
Solution: Correct answer is ‘b’ i.e., Cori cycle
The process of reconversion of lactate formed in the muscles and RBCs, back into glucose inside the liver is called Cori’s cycle.
Lactic acid is a metabolic by-product formed after glycolysis in tissues such as the skeletal muscles and RBCs. The lactate so formed is transported into the liver and kidney. Here, it gets reconverted back into glucose. This entire process is called Cori’s cycle or the glucose – lactate cycle.
Cahill cycle is also called as glucose – alanine cycle. In the fasting state, there is a significant output of alanine from skeletal muscle obtained from glycolysis of muscle glycogen. Alanine is derived from the transamination of pyruvate obtained from glycolysis. Alanine is then exported to the liver where it undergoes transamination again back to pyruvate and enters gluconeogenesis.
Note: Pentose phosphate pathway is otherwise known as Warburg – Dickens – Horecker Cycle.
18: Which of the following enzymes takes part in substrate – level phosphorylation in glycolysis?
a: Pyruvate kinase
b: PFK-1
c: Hexokinase
d: Pyruvate dehydrogenase
Solution: Correct answer is ‘a’ i.e., Pyruvate kinase
The enzyme involved in substrate – level phosphorylation is pyruvate kinase
In substrate level phosphorylation, there is a direct generation of ATP. In glycolysis, there are two such reactions, which are examples of substrate-level phosphorylation. They are as follows:
1: 1, 3 bisphosphoglycerate to 3 – phosphoglycerate, catalyzed by the enzyme phosphoglycerate kinase
2: Phosphoenol pyruvate to pyruvate, catalyzed by pyruvate kinase
19: A 3 year old child presents with muscle weakness, vomiting, and seizures accompanied by focal neurological deficits. Her developmental milestones were normal during the previous visits. Laboratory tests show lactic acidosis and microscopic findings of muscle biopsy are shown below. What is the likely diagnosis?

a: MELAS
b: Duchenne muscular dystrophy
c: Becker muscular dystrophy
d: Myotonic dystrophy
Solution: Correct answer is ‘a’ i.e., MELAS
The given clinical scenario is suggestive of MELAS (mitochondrial encephalopathy, lactic acidosis, and stroke).
It is a mitochondrial inherited condition due to complex I or complex IV deficiency. Patients have normal development previously and then present with symptoms of lactic acidosis and stroke (seizures with focal neurological deficits).
The microscopy of muscle biopsy with modified Gomori trichrome staining shows ragged red fibers (arrowhead) as shown below.
Other mitochondrial inherited diseases:
- Leigh’s disease
- Leber hereditary optic neuropathy
- Kearns – Sayre syndrome
- Chronic progressive external ophthalmoplegia
- Pearson syndrome
20: A TPN bag consists of 100g glucose, 30g amino acids, and 40g lipids. What is the amount of calories delivered?
a: 840 KCal
b: 880 KCal
c: 640 KCal
d: 680 KCal
Solution: Correct answer is ‘b’ i.e., 880 KCal
The amount of calories delivered by the given TPN (Total Parenteral Nutrition) bag is 880 KCal.
The caloric value of different food components are:
- Carbohydrate (glucose) – 4KCal/g
- Proteins (amino acids) – 4.2 KCal/g = ~4KCal/g
- Fat (lipids) – 9 KCal/g
Calculation of the number of calories:
- 100 gm of glucose = 100 x 4KCal = 400 KCal
- 30 gm of amino acids = 30 x 4 KCal = 120 KCal
- 40 gm of lipids = 40 x 9 KCal = 360 KCal
Total amount of calories delivered = 400 + 120 + 360 = 880 KCal
Note: The caloric value of alcohol is 7KCal/g.
21: All of the following enzymes are components of the pyruvate dehydrogenase enzyme complex, except
a: Pyruvate dehydrogenase
b: Dihydrolipoyl transacetylase
c: Pyruvate kinase
d: Dihydrolipoyl dehydrogenase
Solution: Correct answer is ‘c’ i.e., Pyruvate kinase
Pyruvate kinase is not a component of the Pyruvate dehydrogenase (PDH) enzyme complex.
Pyruvate dehydrogenase complex is a multienzyme complex that is associated with the inner mitochondrial membrane which catalyzes the oxidative decarboxylation of pyruvate to Acetyl-CoA.
The three enzyme components are:
1: Pyruvate dehydrogenase
2: Dihydrolipoyl transacetylase
3: Dihydrolipoyl dehydrogenase
The Cofactors are:
1: Thiamine pyrophosphate (TPP)
2: Lipoic acid
3: Coenzyme A
4:Flavin adenine dinucleotide (FAD)
5: Nicotinamide adenine dinucleotide (NAD+)
The overall reaction is: Pyruvate + NAD+ + CoA à Acetyl – CoA + NADH + H+ + CO2
Pyruvate Dehydrogenase is regulated by end-product inhibition & covalent modification. PDH is inhibited by its products, acetyl-CoA, and NADH.
Phosphorylation of the multienzyme complex by a kinase results in decreased activity. The kinase is activated by increases in the [ATP] / [ADP], [acetyl-CoA] [CoA], and [NADH] / [NAD+] ratios.
Dephosphorylation, catalyzed by a phosphatase, causes an increase in activity of PDH. The phosphatase is activated by insulin.
Thus, pyruvate dehydrogenase, and therefore glycolysis, is inhibited when there is adequate ATP available and when fatty acids are being oxidized. In adipose tissue, the enzyme is activated in response to insulin.
Clinical aspects:
1: Arsenite and mercuric ions react with the – SH groups of lipoic acid and inhibit pyruvate dehydrogenase.
2: Dietary deficiency of thiamin inhibits PDH eg. Chronic alcoholics may develop potentially fatal pyruvic and lactic acidosis.
3: Patients with inherited pyruvate dehydrogenase deficiency present with lactic acidosis, particularly after a glucose load and neurological disturbances.
Note: Pyruvate kinase catalyses the last step of glycolysis ie-transfering phosphate group from phosphoenol pyruvate to ADP forming pyruvate and ATP respectively.

Leave feedback about this
You must be logged in to post a comment.